Cystic fibrosis (CF) is a multi-organ life shortening disease typified by chronic endobronchial infection and progressive obstructive lung disease and malnutrition, secondary to pancreatic insufficiency. CF is the most common autosomal recessive condition in Australia, affecting approximately 1 in every 2,500 babies born in Western Australia (WA). Previously a disease of childhood, however, with advances in clinical care, currently there are more adults with CF than children. In the 2022 Australian CF Data Registry, out of a total of 3,738 people with CF, 57% were adults (18+ years).
Currently there is no cure, however, the introduction of mutation-specific therapies and specialised multi-disciplinary treatment, has led to an impressive increase in survival in recent decades. The predicted survival age of a child born after 2017 is 58 years. With newborn screening, many CF centres worldwide are now caring for children with minimal lung damage. It is encouraging that those with CF have the potential to enjoy an increasing life span and an excellent quality of life well into adulthood.
Although life expectancy has improved greatly, the majority of patients still die of respiratory failure. Slowing the progression of lung disease is the primary aim of CF therapy.
Additional CF treatment goals include:
- Reducing early bacterial colonisation
- Slowing airway inflammation to preserve lung function
- Avoiding pulmonary exacerbations
- Optimal nutrition and management of metabolic complications
- Psychosocial support
- Transplantation and appropriate end of life care
The CF Gene
CF results from a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene located on chromosome seven. Over 2,000 CFTR mutations have been discovered; many of these are known to cause CF. The variation in signs and symptoms are associated with the specific CFTR variant or genotype. Genetic background and various environmental factors influence clinical outcome.
In healthy individuals, the CFTR protein forms within the cells then travels to the cell membrane. It then acts as a channel allowing chloride ions to flow out of the cell. Disease causing mutations of the CFTR DNA code result in either a lower amount, or an alteration, that leads to a reduction in the CFTR protein that reaches the cell surface. This CFTR protein reduction means there is decreased chloride secretion and increased sodium absorption in epithelial cells. This causes impaired movement of water in and out of the cells, resulting in abnormal mucus production in the lungs, pancreas, gut and other organs. The alteration in airway surface liquid (ASL), and the resultant increase in viscosity, compromises mucociliary clearance resulting in a predisposition to pathogen colonisation.
The CF Gene Mutation Classification
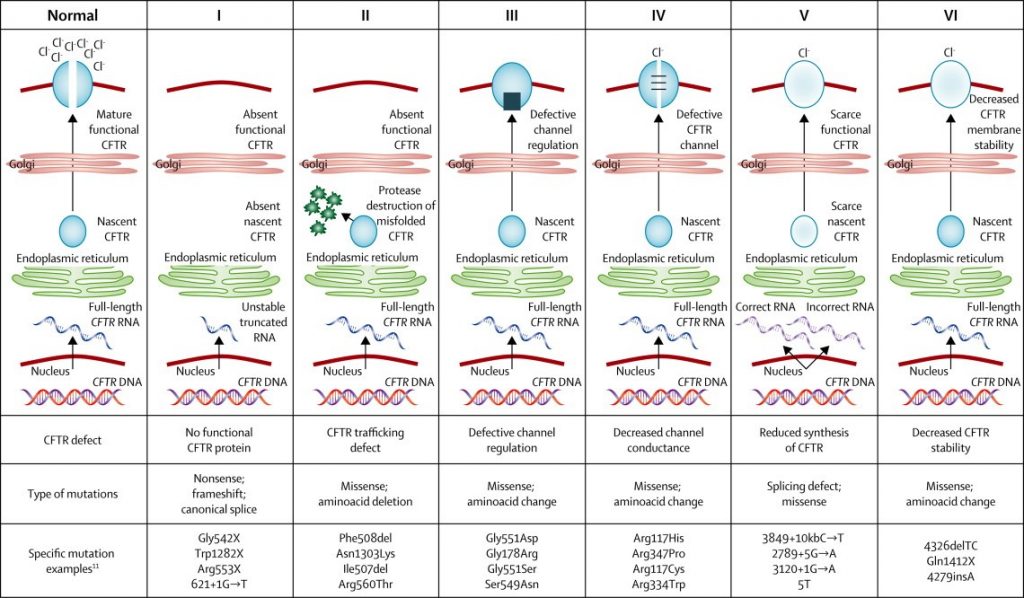
(Boyle & De Boek, 2013)
How CFTR Proteins are Made and What Happens When They Don’t Work Right (Vertex)
Information displayed on vrtx.com is for a US Based audience.
How Certain CFTR Mutations Affect the Way CFTR Proteins Function (Vertex)
Information displayed on vrtx.com is for a US Based audience.
How Certain CFTR Mutations Affect the Quantity of CFTR Proteins at the Cell surface (Vertex)
Information displayed on vrtx.com is for a US Based audience.
Health Professional Resources:
- Understanding CF (Vertex)
- Recognising Disease Progression (CF Source)
- Australian Cystic Fibrosis Data Registry 2022 (Monash University)
Newborn Screening
CF newborn screening (NBS) has been carried out in WA since 2000 and approximately 10-15 babies are born with CF each year. The WA Newborn Screening Program screens for CF along with phenylketonuria, galactosaemia, congenital hypothyroidism, and a range of disorders of amino, organic and fatty acid metabolism.
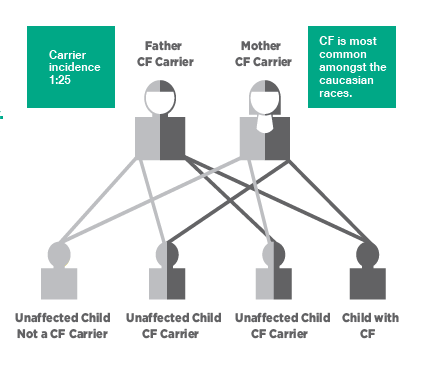
CF is a common inherited disorder with birth prevalence in Australia reported to be approximately 1:3700 (Ruseckaite et al, 2016). Approximately 1 in 25 Australians are carriers of a genetic mutation responsible for CF. Children must inherit 2 defective CFTR genes – one from each parent to have CF. The most commonly known mutation in CF is delta F508.
Newborn screening for CF is a two-step process. It is mainly targeted at detecting elevated levels of immunoreactive trypsinogen (IRT) in the newborn’s blood. Mucus plugging in the pancreatic ducts in a newborn with CF may cause blockages that prevent trypsinogen from reaching the small intestine. This results in an elevated serum trypsinogen. A positive IRT is followed by DNA testing to screen for the most common CF genetic mutations. Infants with one or two common mutations are usually recalled for a sweat test at four to six weeks of age. if only one CF gene is identified they will be a carrier.
Health Professional Resources:
- WA Newborn Screening Program (Department of Health)
Patient Resources:
Carrier Screening
Carrier screening is not the same as newborn screening. Only a very small proportion of CF carriers are identified by newborn screening. It is important for those with a family history of CF to be aware of the options for carrier screening in order to make informed decisions associated with family planning.
Health Professional Resources:
- CF Carrier Screening Program (Cystic Fibrosis Community Care)
- Referral Guidelines for Clinicians (Genetic Services of WA)
- DNA and Relationship Quick Reference Chart (Legacy Tree)
Patient Resources:
Contact Information:
Genetic Services of WA
Obstetrics and General Genetics
Ph: (08) 6458 1525
E: gswa@health.wa.gov.au