Cystic fibrosis (CF) is one of the most common, autosomal recessive, life-limiting diseases affecting children and adults in Australia. It causes thick, sticky mucus which affects the lungs and digestive system.
In the 1950s babies born with CF often died in infancy. However, babies born today can expect to live well into adulthood as CF becomes a more manageable disease due to advances in research and new treatments.
The CF Gene
The gene involved in CF gives instructions for the cells to make a protein that controls the movement of salt in and out of the cells. This salt transport gene lies on chromosome 7. All of us have two copies of chromosome 7 and therefore everyone has two copies of the salt transport gene. Scientists have called this gene the CFTR (Cystic Fibrosis Transmembrane Regulator) gene.
People affected with CF have a defective CFTR gene, causing a malfunction of the movement of salt in and out of the cells lining the exocrine system, which is responsible for producing saliva, sweat, tears and mucus. As a result, there is a constant build-up of excessively thick and sticky mucus within the lungs, airways and the ducts within the digestive system.
Is There a Cure?
At present, there is no cure for CF. Now that the CFTR faulty gene has been identified, global research is moving towards finding ways of repairing or possibly replacing it.
How Do You Get CF?
A child can inherit CF from both parents, getting one abnormal CFTR gene from the mother and another abnormal CFTR gene from the father. Both parents must carry the defective CFTR gene, however the risk of having a child affected with CF is 1 in 4 (or 25%) with each pregnancy.
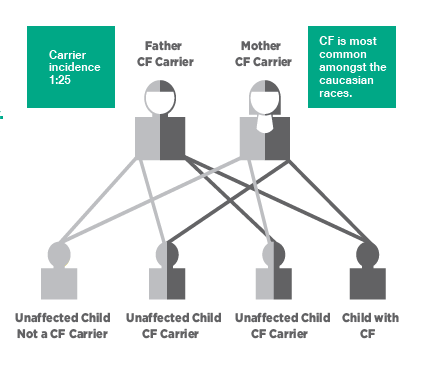
On average, 1 in 25 Australians carry the defective CF gene. However, carriers do not exhibit any symptoms of the genetic disease and most live their lives unaware of being a carrier of the CF gene. In fact, anyone could be a carrier. In Australia about 1 million people are carriers, without being aware of it.
Common Symptoms
The severity of symptoms varies from person to person, and many different factors can affect the course of the disease and the health of a person living with CF.
The Respiratory System
In the respiratory system, thick mucus clogs the airways and traps bacteria, resulting in recurrent infections which cause irreversible lung damage. Lung failure is the major cause of death for someone with CF.
These are some of the common respiratory symptoms:
- Cough productive of mucus
- Recurrent lung and sinus infections
- Wheezing or shortness of breath
The Digestive System
In approximately 85% of people with CF, the digestive system is affected resulting in impaired pancreatic function. Release of enzymes needed for the breakdown of food and the absorption of vital nutrients is inhibited resulting in poor growth and difficulty with weight gain.
These are some of the common digestive symptoms:
- Poor growth or weight gain
- Frequent greasy or oily stools
- Bowel blockages
Other
Other symptoms may include:
- Impaired fertility
- Low bone mineral density
- CF-related diabetes
- Incontinence
- Reflux
Treatment
Airway Clearance
Airway clearance, or chest physiotherapy, is needed to remove thick, sticky mucus from the lungs to prevent lung damage. It is usually started from diagnosis with CF and continues throughout the person’s life.
Nutrition
Diet plays a very important role in the management of CF. In fact, nutrition is so important that a healthy body weight can be linked to better lung function. Pancreatic enzyme replacement therapy is required for people with pancreatic insufficiency.
Medications
Common medications include enzymes, bronchodilators, mucolytics, antibiotics, anti-inflammatories, salt, vitamins and more. Medications may be taken orally, inhaled via a nebuliser, other inhaler device or given intravenously.
Infection Prevention & Control
For people with CF, some bacteria and viruses can cause major lung infections, resulting in hospital admissions and permanent lung damage. Part of everyday life for a person with CF involves reducing the risk of catching an infection, through good hand hygiene practices and avoiding people who are unwell. Individuals with CF also need to avoid contact with other people with CF due to cross-infection of bacteria.