Although a multi-organ disease, it is the pulmonary disease that presents the most challenge and continues to be the main cause of mortality. Most individuals have respiratory disease and exocrine pancreatic insufficiency. The Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) defect is expressed in many epithelial cells including sweat ducts, airway epithelium, pancreatic ducts, intestine, biliary tree and vas deferens.
Respiratory
The chloride and sodium channel defect results in thicker more viscous secretions from exocrine glands, particularly in the respiratory tract, and results in extensive chronic inflammation of the airways. The recurrent lower respiratory tract infections, and a chronic cough with sputum production, leads to structural airway changes including bronchiectasis, obstructive lung disease, and finally, respiratory failure. Bronchiectasis develops in infancy in CF and may be asymptomatic. Studies have shown neutrophilic inflammation and pulmonary infection, especially Pseudomonas aeruginosa (Pa), are major risk factors for early disease in CF, including nutritional and lung function decline (Sly, Gabgell, Chen, Ware, Ranganathan, Scott, & Murray, 2013).
Common symptoms include:
- Cough
- Increased sputum (sputum may be purulent)
- Dyspnoea, hypoxia, tachycardia
- Bronchiectasis
- Haemoptysis
- Lung function decline
- Chest pain
- Pneumothorax
- Sinusitis
- Nasal polyps
- Lethargy/fatigue
- Fever
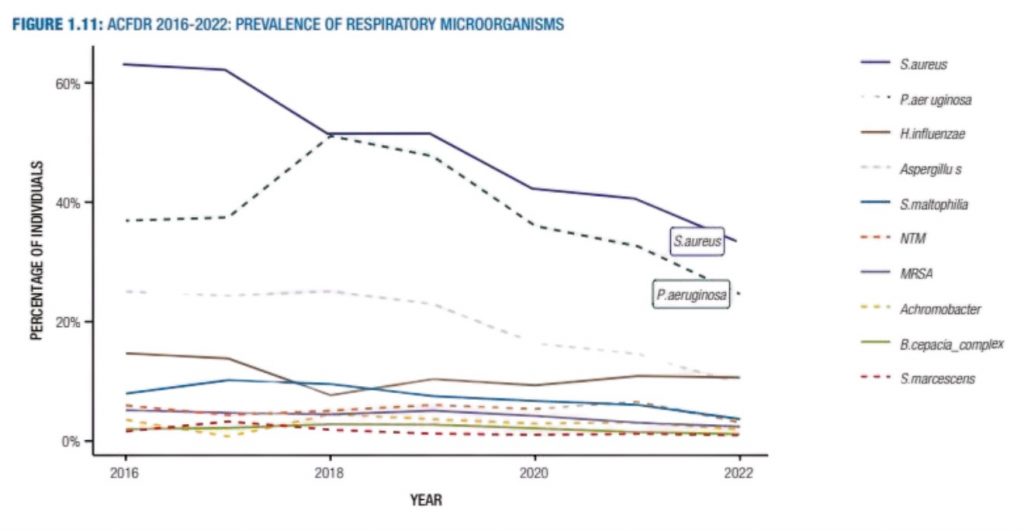
The Australian CF Data Registry 2022 shows there has been a recent reduction in the prevalence of S. aureus, P. aeruginosa and Aspergillus spp since 2016, and most particularly during the last 4 years.
Prevalence of Major Organisms in Lungs
CF care becomes more complex with increasing age and a multidisciplinary health care team is essential. The focus for pulmonary management in CF relates to prevention of airway obstruction by improving mucociliary clearance and prevention and early treatment of respiratory tract infections. A comprehensive approach to maintaining lung health includes airway clearance, physical activity, drug therapy and optimal nutrition.
Airway Clearance
Airway clearance is an important part of CF management and should be used across the lifespan. The physiotherapist’s role involves teaching effective clearance of secretions from the lungs and promoting optimal lung health and general physical fitness. Research has found that no single airway clearance technique is superior and since CF affects each person differently, a treatment program is designed specifically for the individual, and needs to be reviewed regularly. Regular contact with the physiotherapist can help identify small changes, which may be hard for the individual to detect, thus allowing adjustments to the treatment program.
Physiotherapy treatment should be increased when a lung infection occurs and may be more effective when combined with inhaled medications and exercise (Button et al, 2016).

- Modified Postural Drainage (MPD): For babies and young children, when active participation is not possible, the basic program usually involves daily or twice daily modified postural drainage (MPD) with percussion. MPD involves positioning without the use of a head-down tilt. As children grow, physio ‘play’, blowing games, coughing and deep breathing exercises are incorporated into the treatment.
- Active Cycle of Breathing Technique (ACBT): From about three to five years of age more emphasis is placed on breathing exercises. The active cycle of breathing techniques (ACBT) which incorporates deep breaths, breathing control and the forced expiration technique (FET), is taught as a method of clearing mucus more independently. Studies have shown it to be an effective method of mobilising and clearing secretions.
- Positive Expiratory Pressure (PEP): Devices such as Positive Expiratory Pressure (PEP) mask, mouthpiece PEP, bottle PEP, Flutter or Acapella may be prescribed by the physiotherapist to enhance airway clearance. PEP therapy is thought to promote collateral ventilation, increase functional residual capacity and recruit obstructed or collapsed airways.
- Autogenic Drainage (AD): Autogenic Drainage (AD), or self-drainage, is an airway clearance technique that is widely used throughout Europe. The technique is based on the principle of reaching the highest possible airflow in different generations of bronchi by controlling breathing. When performing AD, individuals adjust the rate, depth and location of respiration in order to mobilise secretions.
Health Professional Resources:
- Physiotherapy Clinical Practice Guidelines (The Thoracic Society of Australia and NZ)
- Risk Factors for Bronchiectasis in Children with CF (Sly, et al)
- Australian CF Data Registry 2022 (Monash University)
Patient Resources:
Physical Activity
Another very important early recommendation for airway maintenance is regular exercise. Physical exercise that increases minute ventilation leads to the mobilisation of secretions and enhances airway clearance. It is recommended that all patients should be encouraged to exercise several times a week.
Patient Resources:
Drug Therapy
Managing CF is complex and drug therapy is an important part of CF treatment. Medications are used to ease symptoms, reduce complications and improve quality of life. They may be inhaled, oral or given parentally. A major achievement in treatment are medications that target gene mutations known as CFTR modulator therapies.
Central Vascular Access Devices
Those with CF may require long term antibiotics to treat respiratory exacerbations. Peripherally Inserted Central Catheters (PICC) are used when vascular access is required for more than a week. Infusaports are used for long term access.
Patient Resources:
Mucolytics
Inhaled mucolytic agents are often used as an adjunct to airway clearance techniques and allow direct deposition of drugs to reduce mucus viscosity.
- Dornase alpha (Pulmozyme®): Is a recombinant human deoxyribonuclease (rhDNase). In those with CF, Dornase alpha hydrolyses the DNA of sputum and reduces the viscosity, enhancing airway clearance with the potential to increase lung function and reduce exacerbations.
- Hydrator Therapy: Hydrator therapies are used to restore airway surface liquid hydration by osmotically drawing liquid into airways. Bronchitol® and Hypertonic Saline (HTS) are commonly used in CF. Most patients will require a bronchodilator, such as Salbutamol®, before inhaling Bronchitol® or Hypertonic Saline (HTS).
- Bronchitol® is a form of mannitol, a naturally occurring osmotic agent. It is delivered via a dry powder inhaler prior to airway clearance.
- Inhaled HTS has been shown to increase mucociliary clearance and improve clinical outcomes in children and adults with CF. It is recommended that HTS is administered via a nebuliser before or during airway clearance.
Antimicrobial Therapy
Antimicrobial therapy has been strongly associated with increased survival in those with CF. Long-term use of antibiotics has been significant in slowing lung decline. Antibiotics in CF can be administered orally, by inhalation or intravenously. Commonly used antibiotics may include amoxycillin and clavulanic acid (Augmentin Duo®), flucloxacillin, ciprofloxacin and tobramycin.
Azithromycin is often prescribed for children in subtherapeutic doses. These macrolides are thought to suppress proinflammatory cytokines and reduce the neutrophil burden on the lungs.
Modulator Therapy
CFTR modulators are a new drug therapy that differ from other CF therapies as they aim to improve or restore the function of the defective CFTR protein, rather than offer symptomatic treatment. These therapies are mutation specific. The individual modulators act in different ways to improve the mutant protein function by potentiating action at the cell surface (increasing CFTR channel opening) or correcting the defect by increasing the amount of CFTR protein at the cell surface. Work continues on new modulators in an effort to find an effective treatment for all people with CF.
Currently in Australia the CF community have access to Kalydeco®, Orkambi®, Symdeko® and Trikafta® with many new modulator therapies emerging.
Health Professional Resources:
Patient Resources:
Respiratory Complications
Whilst progressive bronchiectasis and recurrent pulmonary exacerbations are typical of CF lung disease, additional respiratory complications may also occur.
Pneumothorax
Pneumothorax is defined as the presence of air or gas in the space between the visceral and parietal pleura of the lung, which can impair oxygenation and/or ventilation. A sudden onset of breathlessness in patients with CF should be investigated promptly to identify the possibility of a pneumothorax. In more severe cases, hypoxemia and hypercapnia may be observed (Ronan, Elborn & Plant, 2017). In CF, pneumothorax is a marker of disease severity and often associated with lower FEV1, pancreatic insufficiency, haemoptysis and infection. While a small pneumothorax can be managed conservatively, a larger pneumothorax may require surgical intervention.
Haemoptysis
Haemoptysis is defined as the expectoration of blood, or blood-stained mucus, from the lower respiratory tract. Mild to moderate haemoptysis may be an indicator of infective exacerbation and is generally treated conservatively, withholding some of the usual CF treatments and adding antibiotics to treat infections. Treatment of moderate haemoptysis may also include tranexamic acid. Massive haemoptysis may be associated with a positive sputum culture for Staphylococcus aureus and cystic fibrosis related diabetes (CFRD). It is life-threatening and often requires surgical management (Ronan et al, 2017).
Health Professional Resources:
- Current and Emerging Comorbidities in CF (Ronan, et al)
Patient Resources:
Gastrointestinal
Gastrointestinal tract manifestations of CF are related to mucus, dehydration, viscosity and dysmotility.
Common symptoms include:
- Pancreatic insufficiency
- Loss of appetite/weight loss
- Abdominal pain
- Gastro-oesophageal reflux disease (GORD)
- Pancreatitis
- CF related diabetes (CFRD)
- Rectal prolapse
- Distal intestinal obstruction syndrome (DIOS)
- Cirrhosis and other hepatic dysfunction
- Cholelithiasis
- Dehydration
Patients with CF may present at birth with meconium ileus and have higher incidence of complete or partial bowel obstruction, commonly known as DIOS. They may also be more prone to constipation, gastro-esophageal reflux disease, and infections such as C.Difficile. Approximately 90% of Australian CF patients are pancreatic insufficient with consequential poor weight gain, malabsorption of fat and fat-soluble vitamins (Ronan et al, 2017).
Lung function and nutritional status are closely linked. Malnutrition related to inadequate energy intake is a common problem in CF patients. Interdisciplinary management is essential with involvement of a gastroenterologist, endocrinologist and dietitian to manage gastrointestinal (GI) co-morbidities.
Pancreatic Insufficiency
Pancreatic insufficiency refers to significant impairment of the pancreas to secrete sufficient enzymes needed for normal digestion (Saxby, Painter, Kench, King, Crowder, van der Haak, 2017). In individuals with CF, the exocrine glands in the pancreas produce such thick secretions that the pancreatic ducts become blocked. Destruction of acinar pancreatic tissues occurs and lack of enzyme activity results in malabsorption, particularly of fatty foods and fat-soluble vitamins.
The resultant malnutrition and poor growth associated with fat malabsorption and pancreatic insufficiency is often accompanied by steatorrhea, abdominal pain and failure to thrive. With CFTR modulator therapy, the emergence of the overweight CF patient is an increasing phenomenon. It is important patients are screened for undernutrition and overnutrition.
Nutritional Recommendations
Individuals with CF have greater nutritional needs, in particular energy (or calories), compared to the general populations. This is due to the high amount of energy the body uses to breathe, cough, fight infection in the lungs and poor fat digestion. The current guidelines estimate that the energy needs for individuals with CF are 110-200% higher than the general population, however an individual approach must be considered.
To meet these increased nutritional needs, individuals with CF are generally recommended to eat a diet high in energy (or calories). It is important individuals are guided by a CF dietitian as there is not a ‘one-size-fits-all’ method for determining energy needs.
It is important to follow a balanced diet including all food groups (unless otherwise advised) such as grains and cereals, fruits and vegetables, dairy products and protein foods like meat, chicken, fish, legumes, nuts and eggs.
Recommendations often include:
- Pancreatic enzyme replacement therapy (PERT) for those with pancreatic insufficiency
- High energy/high fat diet to optimise weight for those undernourished (healthy fats are encouraged)
- Sodium supplementation
- Supplementation with fat soluble vitamins (ABDECK®)
- Nutritional supplements may be required to optimise nutritional status
- Enteral feeding if indicated
- Use of proton pump inhibitors (PPI) for the treatment of gastro-oesophageal reflux disease (GORD)
- Behavioural therapy to achieve positive meals times
Health Professional Resources:
- Nutrition Guidelines for Cystic Fibrosis (The Thoracic Society of Australia and NZ)
Patient Resources:
Pancreatic Enzyme Replacement Therapy (PERT)
Pancreatic enzyme replacement therapy (PERT), along with vitamin supplementation, is an essential part of the management of pancreatic insufficiency in CF (Saxby et al, 2017). It is important pancreatic enzyme replacement capsules are taken with the first mouthful of foods that contain fat, carbohydrates and protein. The enzyme dosage is related to the fat content of food ingested and an additional dose may be needed if the meal lasts more than 30 minutes.
Creon® is a commonly used PERT in CF. It has a slow release formula designed to deliver enzymes to the duodenum. It can be given as granules mixed with an acidic fruit puree in infants and small children or taken as capsules in older children and adults. PERT adequacy is assessed clinically by monitoring nutritional status, signs of malabsorption and weight gain.
Patient Resources:
Supplements
Vitamins
Those with CF, especially the pancreatic insufficient, are at risk of fat-soluble vitamin deficiencies. VitABDECK® is a CF specific multivitamin for routine supplementation.
Sodium
The CFTR defect results in abolition of normal chloride conductance with consequently poor sodium reabsorption into the cells and a high concentration of salt in sweat. This increased loss of sodium and chloride with exercise and in hot temperatures, predisposes the individual with CF to salt depletion and dehydration. Sodium supplementation is recommended in Australia.
Enteral Feeding and Nutritional Supplements
Enteral feeding can be an effective way to optimise nutrition in those not meeting nutritional requirements. Low profile Percutaneous Endoscopic Gastrostomy (PEG) tubes are often used in children. Intermittent nasogastric tubes may be used by those who would rather a less invasive approach. The CF dietitian will prescribe the appropriate nutritional supplements.
Patient Resources:
Gastrointestinal Complications
The most commonly presenting gastrointestinal complications are cystic fibrosis related diabetes (CFRD) gastro-oesophageal reflux disease, liver cirrhosis, portal hypertension, pancreatitis, cholelithiasis, constipation, distal intestinal obstruction syndrome (DIOS) and rectal prolapse. Lack of functional CFTR in biliary epithelium results in increased viscosity of bile and may lead to liver complications.
Cystic Fibrosis Related Diabetes (CFRD)
The prevalence of CFRD increases with age and is becoming one of the most common co-morbidities associated with CF. It is linked with decreasing lung function, poor nutrition and increased respiratory exacerbations. Acinar atrophy, fatty infiltration and pancreatic fibrosis occur with advancing age and results in decreases in insulin producing beta cells (Ornstein, Rosentstein & Stern, 2000). CFRD affects approximately 20% of adolescent patients and 40-50% of adults (Ronan et al, 2017).
Although CFRD shares similar characteristics with type 1 and type 2 diabetes, it is managed with a typical CF, high energy diet and insulin. All CF patients should be screened for diabetes annually from 10 years of age.
Health Professional Resources:
- Australian Standards of Care for CFRD (2016)
- Current and Emerging Comorbidities in CF (Ronan, et al)
Patient Resources:
Psychosocial
Anxiety and Depression
CF is a chronic life-limiting disease, with a complex and high treatment burden. Anxiety and depression are reported to be two to three times higher for individuals with CF and their carers (Quittner, Abbot, Georgiopoulos, Smith, Hempstead, Marshall, Sabadosa & Elborn, 2016). This can be exacerbated at times of diagnosis, hospital admission, specific medical interventions or another diagnosis e.g. CFRD. Anxiety and depression are normal at times of stress, however, if untreated can lead to unresolved grief and complex trauma which is associated with poor health literacy, low adherence to treatment regimens and poorer health outcomes.
Impacts of Cystic Fibrosis on Mental Health
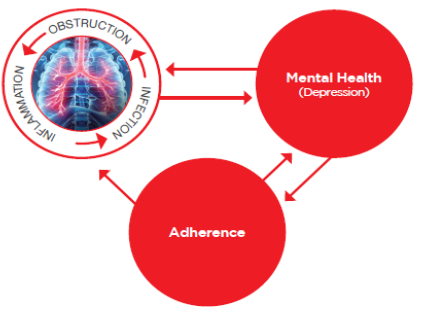
The incidence of depression among children and adults with CF has been reported to be up to 30%. It is recommended that adolescents and adults with CF (ages 12yrs – adulthood) are screened annually for depression and anxiety (Quittner et al, 2016). Living with CF can be emotionally and physically challenging for the patient and family. It is important to recognise and treat anxiety and depressive symptoms in people with CF and their carers, as this can impact on treatment adherence, family functioning, quality of life and has been associated with reduced lung function and increased hospitalisations.
Health Professional Resources:
- International Committee on Mental Health in Cystic Fibrosis (Cystic Fibrosis Foundation and European Cystic Fibrosis Society)
- Chronic Disease Management: Medicare Funded Services (Australian Psychological Society)
Patient Resources (external):
- CF and Mental Health: A Guide for Parents and Caregivers (CFF and CFA)
- Depression, Anxiety and CF: What the Guidelines Mean for You (CFF and CFA)
Pain
Chronic pain is prevalent in adults and children with CF and impacts quality of life and treatment adherence. It has been reported that both adults and children have a high incidence of undertreated pain.
Abdominal pain has been identified as the most prevalent pain issue in 60% of children and 36% of adults. Headaches associated with sinus problems and musculoskeletal pain commonly contribute to a poorer quality of life. Chest pain is often associated with pulmonary exacerbations and can also be associated with physiotherapy. Half of patients with CF do not communicate their pain with a health professional, therefore it is important to include pain assessment as part of the initial assessment (Havermans, Colpaert, Boeck, Dupont & Abbott, 2013).
Pain management goals:
- Improve functional capacity when participating in airway clearance
- Promote lung expansion and better lung function
- Encourage effective mucus clearance
- Reduce stress response and enhances recovery from exacerbations
- Prevent chronic pain and overprotection
Procedural Anxiety
Between 40 and 60% of children report feelings of mild to intense anxiety before an invasive medical treatment. This can also be an ongoing issue for adults.
Children can experience longer term behavioural and psychological issues associated with regular medical procedures. These may include: sleep disturbance, nightmares, separation anxiety, increased irritability, and aggression and temper tantrums. Anxiety or trauma associated with medical treatments can present differently in children depending on their developmental stage. Having an illness or how the illness is perceived can also cause regression to a child’s development.
If a child is experiencing ongoing anxiety and distress associated with their medical treatment, and it can’t be managed with the strategies mentioned above, then it is important to seek professional help. Referral can be made to the Paediatric Consultation Liaison Program, Acute Services Directorate, Perth Children’s Hospital.
Patient Resources:
Other
Cystic fibrosis transmembrane conductance regulator (CFTR) is expressed throughout the entire body resulting in multi-organ manifestations and comorbidities. These may include:
- Bone and join disease including osteopenia and osteoporosis
- Genitourinary disease including infertility in males and stress incontinence
- Kidney disease